
I am pleased to announce the publication, with my friend Etienne Danchin, of an article that criticizes the genetic view of autism and shows why we cannot ignore the crucial role of the environment during pregnancy. In our opinion, the dominance of the genetic view of autism for decades is not only based on questionable grounds but, above all, has not enabled us to better predict, understand or treat autism.
Many brain disorders arise in utero, including autism spectrum disorders (ASDs). They are generated by many intrinsic and extrinsic pathogenic events, including non-functional proteins produced due to genetic mutation, maternal viral or microbial infections, microbiota, epigenetic and immune/ inflammatory signals. Here, we critically review the intrinsic limitations of the solely genomic approach and tools used to assess the genetic origin of ASDs and the underevaluation of environmental hazards during maternity. These observations challenge a direct link between a genetic mutation and the clinical sequelae because of the crucial pathogenic roles of the cascade of alterations produced in utero by the mutation. Major developmental processes including proliferation, migration and synapse formation are gene and activity dependent and are modulated by a wide range of molecular and non-molecular signals. These signals are also vulnerable to environmental insults that can delay/deviate these processes, leading to misconnected/misplaced neuronal ensembles that are the direct final cause of the sequelae and possible therapeutic targets using specific antagonists of these immature patterns. Bringing all the resources and techniques together will require a wide range of scientific approaches, including epidemiology, anatomy, pathology, microbiology, physiology and developmental biology, an interdisciplinary challenge that we must meet if we are to make progress in alleviating and curing disorders born in utero.
Abstract
Brain development involves the sequential expression of vulnerable biological processes including cell proliferation, programmed cell death, neuronal migration, synapse and functional unit formation. All these processes involve gene and activity-dependent events that can be distorted by many extrinsic and intrinsic environmental factors, including stress, microbiota, inflammatory signals, hormonal signals and epigenetic factors, hence leading to disorders born in the womb that are manifested later in autism spectrum disorders (ASDs) and other neurodevelopmental disorders. Predicting and treating such disorders call for a conceptual framework that includes all aspects of developmental biology. Here, taking the high incidence of ASDs as an example, we first discuss the intrinsic limitations of the genetic approach, notably the widely used twin studies and SNPs. We then review the long list of in utero events that can deviate developmental sequences, leading to persistent aberrant activity generated by immature misplaced and misconnected neuronal ensembles that are the direct cause of ASD. In a clinical perspective, we suggest analysing non-genetic maternity data to enable an early prediction of babies who will develop ASD years later, thereby facilitating early psycho-educative techniques. Subsequently, agents capable of selectively silencing malformed immature networks offer promising therapeutic perspectives. In summary, understanding developmental processes is critical to predicting, understanding and treating ASD, as well as most other disorders that arise in the womb.
Introduction
The extraordinary and rapid technological development of new genetic tools at the turn of the third millennium marked the beginning of a new era in the biological sciences, conducive to a better understanding and treatment of diseases. Several projects aimed to define a set of biallelic markers, SNPs capturing most of the common genomic variation to perform genome-wide association studies (GWAS) that are now considered the best way to dissect human diseases. Reviews regularly present in isolation that GWAS have revealed common DNA variations that affect risks of common diseases.1
However, heritabilities derived from GWAS are surprisingly low2 because the major assumptions required to use GWAS are never met in human studies: (1) the disease must be homogeneous, (2) generated by many independent genetic factors, (3) each endowed with small effects and that (4) must not interact with the environment. These conditions are never met in human studies, casting serious doubt on their use in medicine.3 Therefore, a complete paradigm shift is required to better understand and treat disorders.
We review the evidence challenging the validity of a purely genetic approach to autism spectrum disorders (ASDs), which are highly heterogeneous with diagnoses based on deficits of social communication and repetitive behaviours and many sensory, motor, gastrointestinal, immunological alterations, anxiety disorders, intellectual disability, mitochondrial disorders and epilepsy, substantially affecting health, education, social care, welfare, etc.4 Such heterogeneity rules out the assumption of the simple genotype-phenotype relationship used for heritability estimation, explaining why the currently available genomic information remains too poor to identify, let alone alleviate the impairments of, ASD in clinical practice (reviewed in Puricelli et al5).
Genetic and twin studies in ASD
Despite intense search, the genetic interpretation for most forms of ASD remains questionable. For instance, a large-scale GWAS meta-analysis of 18 381 patients with ASD and 27 969 controls disappointingly identified a mere five genome-wide significant loci.6 Even adding seven SNPs detected in a multi-trait GWAS falls short of explaining the full heritability. Therefore, heritability estimates using these techniques ranging from 17% to 91%7 (reviewed in previous work8) are highly controversial because none of the assumptions of the statistical models are met in these analyses. Furthermore, 80.3% of the (n=218) patients with ASD tested in another study did not have any identifiable genetic mutation, and positive de novo variants were observed in 6.9% of patients and depended on parental age.9
The monozygotic (MZ) and dizygotic (DZ) twin comparison presented as an alternative-complementary approach to determine ASD heritability also suffers from intrinsic limitations. MZ twins are the result of a single fertilised egg, whereas DZ twins are the result of two eggs each fertilised by different sperms. A discordance for a trait in MZ twins is interpreted as resulting from different environmental factors, whereas a higher concordance rate in MZ twins than in DZ twins is interpreted as resulting from genes, ignoring environmental factors.10 These methods make two major unmet assumptions: (1) MZ twins are genetically identical, while DZ twins share 50% of their genome, and (2) environmental exposure is equivalent for both throughout embryonic development. Assumption (1) that MZ twins share the same genetic composition is, however, challenged by the fact that 15% of de novo mutations are specific to one of the twins.11 Furthermore, MZ twins also share the same placenta most of the time (circa 70%), as well as friends, classes, parental care, etc, to a larger extent than DZ twins, which can only increase their resemblance beyond their genetic similarity.3 Thus, such assumptions ignore the impact of environmental factors,12 which are often denied, leading to overestimated heritabilities. The many placenta-mediated DNA methylation effects13 should make epigenomes far more similar in MZ than DZ twins,14 and indeed, the MZ genomes are identical early in life but become different as they grow older.15 Therefore, despite the large number of rare genetic mutations identified, most children with ASD lack specific genetic diagnosis, challenging their clinical relevance and suggesting moderate genetic heritability with a substantial role of shared environmental components in twins.16
The impact of de novo mutations
De novo mutations constitute the predominant genetic factor identified in ASD17 with complex yet poorly understood mechanisms. In non-familial ASD, de novo point mutations are mainly of paternal origin and positively correlated with parental age,18 which matches the modest but significant increase in children’s ASD risk with parental age.19 20 The fact that these de novo mutations concern many genes (>500 genes) challenges the idea of a direct genotype-phenotype link. Each of these mutations affects a multitude of genes that are involved in basic functional pathways related to synapse development, axon targeting and neuronal motility, illustrating the wide range of effects on ASD development.21 Only a minority of de novo mutations are postzygotic. Generated by a single or several cell lineages, pre-twinning mutations can increase the differences between germline genomes of MZ twins.11 A large study of parental influence on the germline in Iceland describes an unexpectedly large level of sequence diversity linked to complex interactions between age, sex, mutation type and genomic location,9 22 implying a large variety of interacting factors that cannot be readily included in a single genetic model.23 Furthermore, heritability cannot be interpreted as revealing the proportion of DNA sharing when causal variants are de novo mutations3 or involve inherited epigenetic variants (see next section). The need to identify many biomarkers to fully integrate ASDs’ associated multiple phenotypes contrasts with the limited success ‘to identify phenotypic effects of genetic variants of ASD’.24 It is now obvious that autism research is stuck in a corner, as illustrated by the titles of relevant reviews such as ‘Searching for ways out of the autism maze: genetic, epigenetic and environmental clues’,25 or ‘Lost in translation: traversing the complex path from genomics to therapeutics in autism spectrum disorder’.26 In contrast, the claims that ‘ASD has a high genetic predominance’27 or ‘The cause of ASD is largely genetic’19 are debatable.
ASD inheritance and non-DNA sequence transmission
Extensive observations challenge the unspoken assumption of direct links between mutations and clinical sequelae, and all methods to estimate heritability and risk assume that heredity results exclusively from the DNA sequence transmission, hence allowing us to classify a disease as ‘genetic’. However, the last three decades have brought an impressive accumulation of well-documented examples of non-genetic inheritance in yeast, plants, invertebrates and vertebrates,28 mammals and humans, in link notably with pollution, concerning metabolic disorders (eg, diet-related disorders such as diabetes), cardiac, testis and kidney disorders, and many behavioural traits (eg, depression, mental stress, anxiety, fear, addictions).29 30 Main non-genetic transmission channels involve epigenetics and the microbiota, as well as cultural and ecological inheritance. This is attested in a plethora of reviews on the topic.31–34 A generic mechanism of action of environmental effects on the epigenome of both somatic and germline cells explains how environmental factors can alter germline integrity, hence offspring phenotype.35
In this context, autism represents a textbook example of where the assumption that heritability unquestionably means DNA sequence can trap generations of researchers. The race to find the genes involved (online supplemental table) has not led to any conceptual or therapeutic breakthrough. The intrinsic dynamic property of developmental brain networks and the reactive plasticity in response to genetic or environmental pathological events effectively decouple the ASD phenotypes from any genetic inaugurating pathological event. The identification of a synapse-related genetic architecture of ASD7 remains purely correlational, questioning the genotype-phenotype causality and failing to suggest specific therapeutic targets. Interestingly, the number of genes involved in ASD increases exponentially as genetic testing methods improve,27 implying that in the end far more genes than could be reasonably leveraged for biomedical advances may appear to be involved, suggesting that the search for the genes of ASD is in fact vain in a clinical, therapeutic and cognitive perspective.
The contribution of the gut-brain-immune axis
In disorders like ASD that are born in the womb, many extrinsic and intrinsic environmental insults alter the normal developmental sequence. Among these many environmental insults in utero, gut-brain- immune interactions have been directly linked to ASD, including pathogenesis, and are often underestimated (table 1, figure 1 and previous works8 36). In 1998, acknowledging that (1) most children with autism have painful gut dysfunction,5 8 (2) many of them have a history of antibiotic treatment early in life and (3) that many antibiotics do not affect Clostridia, a class of bacteria known to produce neurotoxic metabolites, Bolte suggested that gut microbiota might be involved in autism.37 She tested this unconventional hypothesis by treating her own child with autism with vancomycin, an antibiotic that affects Clostridia, and found that this drastically improved her son’s behaviour, leading her and colleagues to test on a larger panel, which confirmed her intuition.38 Since then, high-throughput sequencing has greatly improved our ability to study our microbiota, providing a strong case for gut microbiota involvement in ASD (reviewed in table 1). For instance, it was shown that the gut microbiota impacts mammalian brain development and subsequent adult behaviour.39 Also, transplanting the microbiota of autistic humans to germ-free mice led to ‘hallmark autistic behaviours’.40 Furthermore, two papers on the same sample of 18 patients showed that microbiota transfer therapy from neurotypical individuals to patients with autism led to strong improvements that persisted for 8 weeks after transfer, and 2 years later, all symptoms and behaviours were still improving.41 This study not only opens the door for the first time to the possibility of treating patients with ASD in the long term but also shows once and for all that the microbiota plays a prominent role in ASD. It is conceivable that, at least in part, ASD heritability results from maternal-baby transfer of gut microbiota at birth and through colostrum,42 and from the strong selective action of diet on the microbiota,43 the diet likely itself being heritable through cultural transmission.

In addition, maternal inflammation deserves particular emphasis in ASD pathogenesis. Neuroinflammation is produced during pregnancy by maternal viral or microbial insults, immune insults, fever, cardiovascular disorders, obesity, autoimmune disease and psychosocial stress produced by separation from the mother just after birth.44–47 Maternal immune activation provides a causal link leading to neuronal dysfunctions and behavioural phenotypes years or decades later in the adult or aged offspring.48 Prenatal infection of pregnant rodents with influenza virus generates long-term functional, behavioural and brain structural changes in the offspring like those observed in patients.49 Maternal use of the anti-epileptic agent sodium valproate also produces ASD in animal models and humans.50 51 Prenatal pesticides increase the incidence of preterm delivery52 and the incidence of ASD.53 54 Legal or illegal drug use during pregnancy also affects brain development and leads to brain disorders.55 The underlying mechanism link includes early immune activation, inflammatory signals involving interleukins and other signals that derail essential developmental processes including cell proliferation and neuronal migration. Importantly, genetic and environmental events can interact sequentially to produce the deleterious effects illustrating the complexity of the disease but also challenging any approach that relies solely on genetic mutations and postulates that there is a direct link between the mutation and the ultimate cause of the sequelae.
Early identification of ASD
Two complementary studies have shown that the impaired in utero developmental processes significantly change the morphological traits of future babies with ASD to the point that they can be identified with reasonable accuracy right at the time of birth.
In the first one, the comparison of late gestation brain growth between fetuses who will or will not later develop ASD showed brain overgrowth starting during the second half of the second trimester of gestation (22nd week of amenorrhea).56 The second more detailed study used the more than 140 parameters routinely collected in French maternities from 63 ASD matched with 189 neurotypic babies born in the same maternity and conditions.57 A machine learning program identified at birth almost half of the future babies with ASD with a high degree of specificity and virtually all neurotypic babies (96%). After validation in many maternities, a test may be developed and applied routinely in maternities to predict babies who will later be diagnosed with ASD. The relevant measurements include expected ones (viral or microbial infections), but also unexpected parameters such as greater femur size during the second trimester, or earlier position face down before birth. Also, the fetal head circumference (HC) is significantly larger during the third trimester of gestation in 38% of the fetuses that will develop ASD compared with age-matched neurotypic fetuses (figure 2).57 This parallels the bigger brain circumference observed in circa 40% of babies and infants with ASD.58
Interestingly, the third ultrasound performed shortly before delivery shows that all ‘future ASD’ fetuses have bigger HC than age-matched neurotypical ones,57 suggesting a deficiency of an essential process that takes place in preparation for birth. In addition, the HC of circa 40% of future children with ASD is larger than that of other fetuses already at the second trimester (figure 2, right panel), suggesting that the larger HC reported in children and adolescents is conspicuous in utero. This has been validated experimentally in rodents, where the volume of the hippocampus and the neocortex are found to be bigger after birth than shortly before in ASD compared with age-matched controls.59 Perhaps more surprisingly, comparing hippocampal neuron size before and after delivery reveals that neurons continue to grow in ASD but not in control pups.59 Therefore, neurons and structure volume increase during birth in ASD but not in controls. Clearly, ASD is born in utero so that developmental sequences are modified, including in relation to the complex events occurring during birth.

This is attested by anatomo-pathological signatures such as patches of cortical malformations and altered cortical columns, changes in numbers of neurons and size of brain structures.60 This calls for establishing reliable diagnostic methods to detect children with autism right at birth. Yet, in most developed countries, despite recommendations from both medical academies and political authorities, diagnosis is established only around 4–5 years, resulting in an apparent age of the onset of the developmental disorder around 18 months. Late detection misses the best window of opportunity for treatment and leads to a lifetime of disability that could have been substantially mitigated by early treatment (figure 3). The importance of early detection and intervention in curing children with ASD has been discussed thoroughly elsewhere.61

Critical periods and temporality of developmental insults
In brain development, “the only constant is change” (Ephesus, c. 535–475 BC). Major modifications occur in the developing brain. Several essential steps, including cell proliferation, migration, synapse and neuronal ensemble formation, are largely unique to the developing brain. Virtually all ionic currents, including voltage or transmitter-gated currents, and network activities sharply differ and exert different functions in the developing and adult brain.62–65 In summary, the developing brain is not a small adult brain but one endowed with highly specific processes that must take place timely.
Moreover, brain development is not a continuous linear process as it involves many ‘critical periods’ representing milestones in the establishment of functional entities. Altering these critical periods leads to severe sequelae in humans and animals.66 For instance, migration disorders produced by genetic mutations or environmental and inflammatory insults during the cortical migration period are a leading cause of drug-resistant epilepsies and intellectual disability.60 67 Similarly, insults that occur during the critical period of the visual system impact its development.68 In the preterm fetus, spontaneous movements are associated with electrical activity that propagates from the periphery to the centre, acting as a sensory feedback system that controls the formation of cortical maps.69 Drug treatments or behavioural manipulations like pharmacological interventions can advance or delay these critical periods with long-term consequences.70
Parturition and birth are also major critical periods. The ‘stress of being born’ is associated with massive release of stress hormones needed for respiratory functions71 but potentially toxic for neuronal activity. Oxytocin, which triggers labour, also attenuates the effects of stress hormones exerting analgesic actions.72 73 Interestingly, fetal HC just before birth is larger in future children with ASD than in age-matched neurotypical children, suggesting that a protective event prior to birth was abolished in ASD (figure 2). In keeping with this, in rodent ASD models, the volume of the hippocampus and hippocampal neurons grows during birth but not in age-matched controls.59 Preterm and, to a lesser extent, C-section delivery are associated with increased incidence of ASD.74 75 The underlying mechanisms, including inflammatory signals in utero, must be considered in models aimed at explaining the pathogenesis of ASD.
Lost in translation: the purely genomic approach is a
therapeutic dead end
The primary goal of applying genomic tools to ASD was to better understand the pathogenic processes and suggest new therapeutic avenues for ASD. Despite considerable efforts, these targets have not been met26 76 even for well-characterised single gene mutation like Fragile X (FRX). Indeed, as a single gene disorder, it was thought that FRX would have consistent drug targets that could be modulated with various drugs. However, despite promising experimental observations, translational drug treatment has largely failed.77 The reasons for these failures are the clinical heterogeneity of FRX calling for the identification of subpopulations responding to the treatment.78 In addition, the single mutations associated with FRX (or Rett syndrome) are commonly associated with ASD and have complex targets with multiple cascades of alteration already in utero, suggesting multiple series of alterations early on and precluding considering a direct relation between a single mutation and a syndrome. However, more recently, a preliminary trial using an inhibitor of phosphodiesterase (BPN 14770) showed promising effects on children with FRX,79 although the heterogeneity of FRX might be unravelled later in larger trials as in ASD.
It is important to stress the many natural and important shifts occurring during brain development. All ionic currents and network-driven patterns differ in immature and adult brains, exerting different roles. In the developing brain, neuronal activity modulates neuronal proliferation, migration, synapse formation and even cell phenotype: in amphibia spinal cord, the GABA or glutamate phenotype is determined by ongoing activity. The checkpoint concept posits that developing network activity and gene expression constitute obligatory gateways that control the timing and activation of essential developmental processes.80 Altering or impacting this sequence has major long-lasting sequelae. Thus, increasing calcium spike frequency in excitatory glutamatergic neurons shifts them to become GABAergic inhibitory and conversely for GABAergic neurons, thereby preserving an excitatory/inhibitory balance (figure 4, left panel).81 The most investigated developmental shift concerns the GABA excitatory to inhibitory developmental shift that has been preserved throughout evolution. GABA exerts a wide range of trophic actions including on cell growth, synapse and network formation.63 This developmental shift is due to a progressive reduction of (Cl-)i levels that are elevated in young neurons and low in adults63 (figure 4, right panel). This developmental alteration is due to the enhanced activity of the NKCC1 chloride importer in immature neurons.63 82 This developmental sequence generates a variety of patterns that are unique to the developing brain in rodents83 and non-human primates84 with equivalent patterns in human activity.69 85 Interestingly, pathological events occurring during such a dynamic process perturb these checkpoints, leading to permanent alterations including misplaced or misconnected neuronal ensembles. In keeping with this, in a wide range of disorders, including ASDs, various epilepsies, Fragile X, Down syndrome, etc, GABA excites adult neurons consequently to exacerbated NKCC1 activity, contributing to the sequelae. The Neuroarchaeology concept posits that these persistent alterations are the final cause of disorders born in utero and possible therapeutic targets using selective inhibitors of these immature features.86
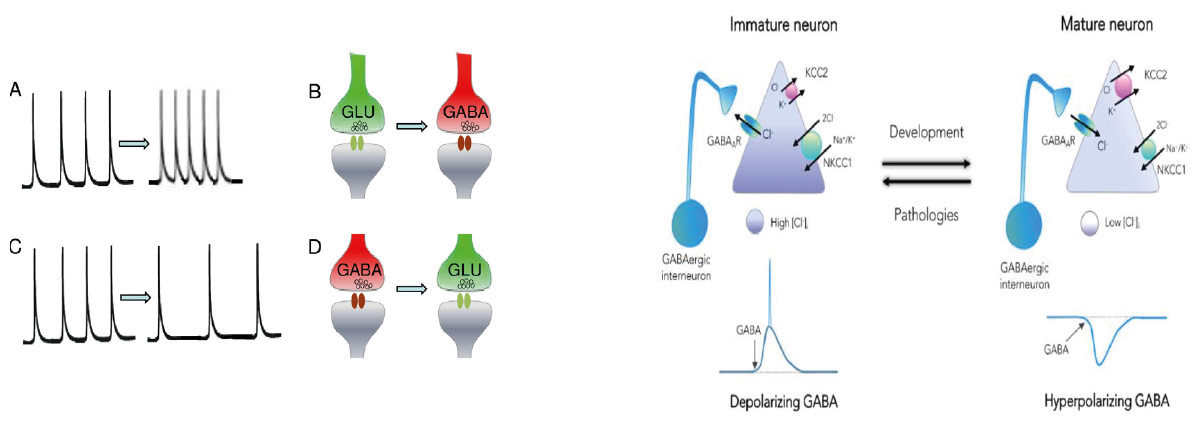
Relying on the enhanced NKCC1 activity, high (Cl-)i levels and excitatory actions of GABA on models of ASD,72 several trials have tested the effects of a selective inhibitor of NKCC1 chloride importer to attenuate GABA excitatory actions. The inhibitor of NKCC1 chloride importer bumetanide has been shown to restore GABAergic inhibition in various pathological animal models. A monocentric87 followed by a multicentric88 phase II double-blind, randomised trials showed a statistically significant difference between placebo and bumetanide-treated children. Using the same conditions and dosage, similar phase II trials were successfully conducted in China (three trials), Holland, Egypt, Tunisia, with a total of over 1036 children treated according to a meta-analysis. 89 Eye tracking measures and brain imaging showed, respectively, a significant amelioration of visual contact and reduced activation of the amygdala in response to constrained eye-to- eye communication.90 91 Unfortunately, two final phase III trials performed in many European countries, Brazil and Australia (210 children 2–6 years old and 210 children aged 7–18 years old) failed to observe a significant difference between children treated with placebo and bumetanide using CARS2 to evaluate attenuation.92 This is in line with the repeated failure of large phase III trials in ASD, Fragile X and other neurodevelopmental disorders even despite promising phase II trials. This is most likely due to the heterogeneity of ASD and the need to identify subpopulations and better stratify inclusion criteria relying on clinical or biological markers. This suggests that a single treatment for all ASD phenotypes is unlikely. The recent demonstration that EEG parameters enable to identify adolescent’s responder to bumetanide versus non-responders provides an illustration of this novel avenue.93 In other words, future development of ASD treatments is conditioned by the clinical identification of homogeneous subpopulations of children with ASD using large analysis facilities and notably machine learning. Although illustrated with the GABA excitatory-inhibitory shift, many ionic current and other biological or biochemical candidates may be found in the future to be involved in those disorders. This situation is reminiscent of the ‘foetal origin of disorders’ developed for diabetes and cardiovascular diseases, which was shown to be quite useful to treat disorders born in the womb.94
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Our review leads us to propose a therapeutic strategy (figure 3). ASDs, like many other neurodevelopmental disorders, are vastly heterogeneous thereby ‘tempering hope for momentous aetiological discoveries in the near future that could tractably pinpoint specific, mechanistic and curative treatments to these conditions’. 61 Far from being primus inter pares, genetic information is but one of many factors that regulate brain development, and therefore, the treatment of ASDs and other neurodevelopmental disorders depends on a better understanding of their developmental pathogenesis. Disorders born in utero, such as ASDs, require a complete methodological shift from a purely genomic approach to one that includes a wide range of expertise.
The key step in this shift is to detect disorders at the earliest possible stage to use the crucial developmental period of exceptional plasticity during the years following birth to at least alleviate most impairments of ASD (figure 3). For instance, the benefit of early diagnosis was demonstrated in congenital hearing or visual problems.95 However, and very sadly, over 50% of patients are only identified at school because of their severe disruptive behaviour and poor social and communicative abilities.61
Early identification of ASD also implies redefining the sequence of early brain developmental stages and checkpoints.61 We posit that the heterogeneity of ASDs is the by-product of the different timing of occurrence of the initial genetic-environmental insult. Thus, the identification and validation of biomarkers of ASDs (such as HC) must drive the collection of developmental data in maternities and should be part of the routine monitoring protocols of pregnant women to allow their generalised use to detect and treat future patients with ASD right at birth and use psycho-educative techniques to reinforce social interactions. Admittedly, this task implies changing the monitoring protocols of pregnant women in several countries, but this should not be a major difficulty in view of the potential major benefits. Furthermore, the large collection of this type of data will in turn help improve our understanding of the heterogeneity of ASDs. Such a routine protocol can also be complemented by gathering epigenetic data in specific subsamples as epigenetics clearly appear to be involved in ASD,96 as well as any other marker that might be identified as associated with ASD in the future.
It is now crucial to replace the idea that ASD is a ‘genetic’ disease by the more accurate idea that ASD is a ‘multifactorial-syndrome- with- a- possible- genetic- risk’. This will also shift our efforts from ‘curing the disease’ to ‘optimising outcomes’ to better understand and attenuate the deleterious sequelae.61 In fact, the purpose of a treatment is not to cure a patient of a constellation of traits but rather to ensure that inborn deficiencies do not translate into permanent disabilities.61 This conceptual shift will also reflect the realisation that ASDs and other neurodevelopmental disorders are ‘extreme ends of continuously distributed dimensions, akin to hypertension along the continuum of blood pressure’.61 In a therapeutic perspective, the Neuroarchaeology concept should help identify persistent immature features expressed in experimental conditions that are amenable to being silenced by selective drugs. The GABA excitatory shift with the NKCC1 inhibitor is but one example of this approach. The identification of the aberrant networks generated by the inaugurating insult should help develop drugs that would specifically silence these ‘immature’ networks.
Furthermore, it should be emphasised that although we have reviewed data relevant to ASD, our conclusions are valid for many other neurodevelopmental disorders, including some of genetic origin associated with ASD-type symptoms. For example, the depolarising effect of GABA leading to a shift of the excitatory/ inhibitory balance, as well as the overactivity of NKCC1 over Kcc2 and positive effects of NKCC1, has been reported in Fragile X,85 Down syndrome,97 Rett syndrome and 22q11-2 deletion syndrome.98–100 For all of them, similar potential therapeutics using bumetanide and other strategies aimed at reducing NKCC1 activity or enhancing the activity of the exporter KCC2 have also been suggested.85 97–100 Therefore, our conclusions have a much broader scope than the sole application to ASD, which here is used as an example of the general need to integrate all forms of inheritance into our medical approaches.
In conclusion, we urge researchers and practitioners to move beyond the mainstream reductive deterministic view of life that focuses too much on DNA sequence and to adopt an approach that integrates information coming from all areas of biology. This approach should consider the temporality of the onset of disorders and the dynamic basic property of brain networks to be able to detect and act before irreversible damage is done. Bringing all the resources and techniques together will require drawing on numerous scientific approaches, including epidemiology, anatomy, pathology, microbiology, physiology and developmental biology, an interdisciplinary challenge that we must meet if we are to make progress in alleviating and curing our many complex diseases.
SOURCES
1 Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 2017;101:5–22.
2 Maher B. Personal genomes: The case of the missing heritability. Nature New Biol 2008;456:18–21.
3 Robette N, Génin E, Clerget-Darpoux F. Heritability: What’s the point? What is it not for? A human genetics perspective. Genetica 2022;150:199–208.
4 Hodges H, Fealko C, Soares N. Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Transl Pediatr 2020;9:S55–65.
5 Puricelli C, Rolla R, Gigliotti L, et al. The Gut-Brain- Immune Axis in Autism Spectrum Disorders: A State-of- Art Report. Front Psychiatry 2021;12:755171.
6 Grove J, Ripke S, Als TD, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019;51:431–44.
7 Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 2015;16:551–63.
8 Nitschke A, Deonandan R, Konkle ATM. The link between autism spectrum disorder and gut microbiota: A scoping review. Autism 2020;24:1328–44.
9 Geier DA, Kern JK, Sykes LK, et al. Examining genotypic variation in autism spectrum disorder and its relationship to parental age and phenotype. Appl Clin Genet 2016;9:121–9.
10 Falconer DS. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genet 1965;29:51–76.
11 Jonsson H, Magnusdottir E, Eggertsson HP, et al. Differences between germline genomes of monozygotic twins. Nat Genet 2021;53:27–34.
12 Bölte S, Girdler S, Marschik PB. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci 2019;76:1275–97.
13 Bahado-Singh RO, Vishweswaraiah S, Aydas B, et al. Artificial intelligence and placental DNA methylation: newborn prediction and molecular mechanisms of autism in preterm children. J Matern Fetal Neonatal Med 2025;35:8150–9.
14 van Dongen J, Gordon SD, McRae AF, et al. Identical twins carry a persistent epigenetic signature of early genome programming. Nat Commun 2021;12:5618.
15 Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005;102:10604–9.
16 Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011;68:1095–102.
17 Weiner DJ, Wigdor EM, Ripke S, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 2017;49:978–85.
18 Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature New Biol 2012;488:471–5.
19 Sandin S, Yip BHK, Yin W, et al. Examining Sex Differences in Autism Heritability. JAMA Psychiatry 2024;81:673.
20 O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature New Biol 2012;485:246–50.
21 Gilman SR, Iossifov I, Levy D, et al. Rare De Novo Variants Associated with Autism Implicate a Large Functional Network of Genes Involved in Formation and Function of Synapses. Neuron 2011;70:898–907.
22 Jónsson H, Sulem P, Kehr B, et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature New Biol 2017;549:519–22.
23 Wroten M, Yoon S, Andrews P, et al. Sharing parental genomes by siblings concordant or discordant for autism. Cell Genom 2023;3:100319.
24 Rolland T, Cliquet F, Anney RJL, et al. Phenotypic effects of genetic variants associated with autism. Nat Med 2023;29:1671–80.
25 Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci 2006;29:349–58.
26 Sestan N, State MW. Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Neuron 2018 ;100:406–23.
27 Kereszturi É. Diversity and Classification of Genetic Variations in Autism Spectrum Disorder. Int J Mol Sci 2023;24:16768.
28 Perez MF, Lehner B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat Cell Biol 2019;21:143–51.
29 Zeid D, Gould TJ. Impact of nicotine, alcohol, and cocaine exposure on germline integrity and epigenome. Neuropharmacology 2020;173.
30 Bohacek J, Mansuy IM. Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet 2015;16:641–52.
31 Wang Y, Liu H, Sun Z. Lamarck rises from his grave: parental environment-induced epigenetic inheritance in model organisms and humans. Biol Rev Camb Philos Soc 2017;92:2084–111.
32 Danchin E, Pocheville A, Rey O, et al. Epigenetically facilitated mutational assimilation: epigenetics as a hub within the inclusive evolutionary synthesis. Biological Rev 2019;94:259–82.
33 Fitz-James MH, Cavalli G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat Rev Genet 2022;23:325–41.
34 Danchin E. From the modern synthesis to the inclusive evolutionary synthesis: an einsteinian revolution in evolution. In: du Crest A, Valković M, Ariew A, et al, eds. Evolutionary thinking across disciplines; problems and perspectives in generalized Darwinism. Switzerland: Springer Nature Switzerland AG, 2023: 401–27.
35 Danchin É, Pocheville A, Huneman P. Early in life effects and heredity: reconciling neo-Darwinism with neo-Lamarckism under the banner of the inclusive evolutionary synthesis. Phil Trans R Soc B 2019;374:20180113.
36 Cryan JF, O’Riordan KJ, Cowan CSM, et al. The Microbiota-Gut- Brain Axis. Physiol Rev 2019;99:1877–2013.
37 Bolte ER. Autism and Clostridium tetani. Med Hypotheses 1998;51:133–44.
38 Sandler RH, Finegold SM, Bolte ER, et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J Child Neurol 2000;15:429–35.
39 Heijtz RD, Wang S, Anuar F, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 2011;108:3047–52.
40 Sharon G, Cruz NJ, Kang D-W, et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019;177:1600–18.
41 Kang D-W, Adams JB, Vargason T, et al. Distinct Fecal and Plasma Metabolites in Children with Autism Spectrum Disorders and Their Modulation after Microbiota Transfer Therapy. mSphere 2020;5:e00314-20.
42 Toquet M, Gómez-Martín Á, Bataller E. Review of the bacterial composition of healthy milk, mastitis milk and colostrum in small ruminants. Res Vet Sci 2021;140:1–5.
43 Salonen A, de Vos WM. Impact of diet on human intestinal microbiota and health. Annu Rev Food Sci Technol 2014;5:239–62.
44 Edlow AG. Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat Diagn 2017;37:95–110.
45 Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci 2017;20:145–55.
46 Han VX, Patel S, Jones HF, et al. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol 2021;17:564–79.
47 Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism 2017;8:13.
48 Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013;155:1451–63.
49 Shi L, Fatemi SH, Sidwell RW, et al. Maternal Influenza Infection Causes Marked Behavioral and Pharmacological Changes in the Offspring. J Neurosci 2003;23:297–302.
50 Christensen J, Grønborg TK, Sørensen MJ, et al. Prenatal Valproate Exposure and Risk of Autism Spectrum Disorders and Childhood Autism. JAMA 2013;309:1696.
51 Ingram JL, Peckham SM, Tisdale B, et al. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol 2000;22:319–24.
52 Kolan AS, Hall JM. Association of Preterm Birth and Exposure to Endocrine Disrupting Chemicals. Int J Mol Sci 2023;24:1952.
53 Shelton JF, Geraghty EM, Tancredi DJ, et al. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: the CHARGE study. Environ Health Perspect 2014;122:1103–9.
54 Mostafalou S, Abdollahi M. Pesticides: an update of human exposure and toxicity. Arch Toxicol 2017;91:549–99.
55 Thompson BL, Levitt P, Stanwood GD. Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat Rev Neurosci 2009;10:303–12.
56 Bonnet-Brilhault F, Rajerison TA, Paillet C, et al. Autism is a prenatal disorder: Evidence from late gestation brain overgrowth. Autism Res 2018;11:1635–42.
57 Caly H, Rabiei H, Coste-Mazeau P, et al. Machine learning analysis of pregnancy data enables early identification of a subpopulation of newborns with ASD. Sci Rep 2021;11:6877.
58 Libero LE, Nordahl CW, Li DD, et al. Persistence of megalencephaly in a subgroup of young boys with autism spectrum disorder. Autism Res 2016;9:1169–82.
59 Cloarec R, Riffault B, Dufour A, et al. Pyramidal neuron growth and increased hippocampal volume during labor and birth in autism. Sci Adv 2019;5:eaav0394.
60 Casanova MF, Buxhoeveden DP, Switala AE, et al. Minicolumnar pathology in autism. Neurology (ECronicon) 2002;58:428–32.
61 Klin A, Micheletti M, Klaiman C, et al. Affording autism an early brain development re-definition. Dev Psychopathol 2020;32:1175–89.
62 Spitzer NC, Borodinsky LN. Implications of activity-dependent neurotransmitter– receptor matching. Phil Trans R Soc B 2008;363:1393–9.
63 Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 2002;3:728–39.
64 Chalupa LM. Retinal waves are unlikely to instruct the formation of eye-specific retinogeniculate projections. Neural Dev 2009;4:1–7.
65 Ben-Ari Y, Gaiarsa J-L, Tyzio R, et al. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 2007;87:1215–84.
66 Desai NS, Cudmore RH, Nelson SB, et al. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 2002;5:783–9.
67 Ackman JB, Aniksztejn L, Crépel V, et al. Abnormal network activity in a targeted genetic model of human double cortex. J Neurosci 2009;29:313–27.
68 Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr Opin Neurobiol 2000;10:138–45.
69 Milh M, Kaminska A, Huon C, et al. Rapid cortical oscillations and early motor activity in premature human neonate. Cereb Cortex 2007;17:1582–94.
70 Marguet SL, Le-Schulte VTQ, Merseburg A, et al. Treatment during a vulnerable developmental period rescues a genetic epilepsy. Nat Med 2015;21:1436–44.
71 Lagercrantz H, Slotkin TA. The “stress” of being born. Sci Am 1986;254:100–7.
72 Tyzio R, Nardou R, Ferrari DC, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 2014;343:675–9.
73 Mazzuca M, Minlebaev M, Shakirzyanova A, et al. Newborn Analgesia Mediated by Oxytocin during Delivery. Front Cell Neurosci 2011;5:3.
74 Curran EA, O’Neill SM, Cryan JF, et al. Research review: Birth by caesarean section and development of autism spectrum disorder and attention-deficit/ hyperactivity disorder: a systematic review and meta-analysis. J Child Psychol Psychiatry 2015;56:500–8.
75 Padilla N, Eklöf E, Mårtensson GE, et al. Poor Brain Growth in Extremely Preterm Neonates Long Before the Onset of Autism Spectrum Disorder Symptoms. Cereb Cortex 2017;27:1245–52.
76 Myers SM, Challman TD, Bernier R, et al. Insufficient Evidence for “Autism-Specific” Genes. Am J Hum Genet 2020;106:587–95.
77 Erickson CA, Davenport MH, Schaefer TL, et al. Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord 2017;9:7:7:.
78 Verdura E, Pérez-Cano L, Sabido-Vera R, et al. Heterogeneity in Fragile X Syndrome Highlights the Need for Precision Medicine-Based Treatments. Front Psychiatry 2021;12:722378.
79 Berry-Kravis EM, Harnett MD, Reines SA, et al. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, phase 2 clinical trial. Nat Med 2021;27:862–70.
80 Ben-Ari Y, Spitzer NC. Phenotypic checkpoints regulate neuronal development. Trends Neurosci 2010;33:485–92.
81 Spitzer NC. Neurotransmitter Switching in the Developing and Adult Brain. Annu Rev Neurosci 2017;40:1–19.
82 Rivera C, Voipio J, Payne JA, et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature New Biol 1999;397:251–5.
83 Leinekugel X, Khazipov R, Cannon R, et al. Correlated bursts of activity in the neonatal hippocampus in vivo. Science 2002;296:2049–52.
84 Khazipov R, Esclapez M, Caillard O, et al. Early development of neuronal activity in the primate hippocampus in utero. J Neurosci 2001;21:9770–81.
85 He Q, Nomura T, Xu J, et al. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci 2014;34:446–50.
86 Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci 2008;31:626–36.
87 Lemonnier E, Degrez C, Phelep M, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry 2012;2:e202.
88 Lemonnier E, Villeneuve N, Sonie S, et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 2017;7:e1056.
89 Xiao H-L, Zhu H, Jing J-Q, et al. Can bumetanide be a miraculous medicine for autism spectrum disorder: Meta-analysis evidence from randomized controlled trials. Res Autism Spectr Disord 2024;114:102363.
90 Hadjikhani N, Åsberg Johnels J, Lassalle A, et al. Bumetanide for autism: more eye contact, less amygdala activation. Sci Rep 2018;8:3602.
91 Hadjikhani N, Zürcher NR, Rogier O, et al. Improving emotional face perception in autism with diuretic bumetanide: a proof-of- concept behavioral and functional brain imaging pilot study. Autism 2015;19:149–57.
92 Fuentes J, Parellada M, Georgoula C, et al. Bumetanide oral solution for the treatment of children and adolescents with autism spectrum disorder: Results from two randomized phase III studies. Autism Res 2023;16:2021–34.
93 Juarez-Martinez EL, van Andel DM, Sprengers JJ, et al. Bumetanide Effects on Resting-State EEG in Tuberous Sclerosis Complex in Relation to Clinical Outcome: An Open-Label Study. Front Neurosci 2022;16:879451.
94 Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 2014;94:1027–76.
95 Korver AMH, Smith RJH, Van Camp G, et al. Congenital hearing loss. Nat Rev Dis Primers 2017;3:16094.
96 LaSalle JM. Epigenomic signatures reveal mechanistic clues and predictive markers for autism spectrum disorder. Mol Psychiatry 2023;28:1890–901.
97 Deidda G, Parrini M, Naskar S, et al. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat Med 2015;21:318–26.
98 Ip JPK, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 2018;19:368–82.
99 Smrt RD, Eaves-Egenes J, Barkho BZ, et al. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis 2007;27:77–89.
100 Amin H, Marinaro F, De Pietri Tonelli D, et al. Developmental excitatory-to- inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci Rep 2017;7:15752.