
J’ai la plaisir de vous annoncer la publication avec mon ami Etienne Danchin d’un article qui critique la vision génétique de l’autisme et montre combien on ne peut ignorer le rôle crucial de l’environnement pendant la maternité. A notre avis, la domination depuis des décennies du champ de l’autisme par la vision génétique a non seulement une base contestable mais surtout n’a permis ni de mieux prédire, comprendre ou traiter l’autisme.
Le développement du cerveau implique l’expression séquentielle de processus biologiques vulnérables, notamment: la prolifération cellulaire, la mort cellulaire programmée, la migration neuronale, la formation de synapses et d’unités fonctionnelles. Tous ces processus impliquent des événements dépendant des gènes et de l’activité qui peuvent être faussés par de nombreux facteurs environnementaux extrinsèques et intrinsèques, notamment le stress, le microbiote, les signaux inflammatoires, les signaux hormonaux et les facteurs épigénétiques, hormonaux et des facteurs épigénétiques, ce qui entraîne des troubles à la naissance qui se manifestent plus tard par des troubles du spectre autistique (TSA) et d’autres troubles neurodéveloppementaux. La prédiction et le Le traitement de ces troubles nécessite un cadre conceptuel qui inclut tous les aspects de la biologie du développement. En prenant l’exemple de l’incidence élevée des TSA, nous discutons tout d’abord des limites intrinsèques de l’approche génétique, notamment le fait qu’il est difficile d’évaluer l’incidence des TSA, et les études de jumeaux et les polymorphismes mononucléotidiques largement utilisés. Nous passons ensuite en revue la longue liste d’événements in utero qui peuvent dévier les séquences de développement et conduire à une activité aberrante persistante générée par le système immunitaire. Cette activité aberrante persistante est générée par des ensembles neuronaux immatures mal placés et mal connectés qui sont la cause directe des TSA. D’un point de vue clinique, nous suggérons d’analyser les données maternelles non génétiques maternelles recueillies normalement dans les maternités permettent une identification précoce des bébés qui développeront un TSA des années plus tard, ce qui faciliterait l’usage des techniques psycho-éducatives précoces. Par la suite, les agents capables de réduire au silence de manière sélective les réseaux immatures malformés offrent des perspectives thérapeutiques prometteuses.En résumé, la compréhension des processus de développement est essentielle pour prédire, comprendre et traiter les TSA, ainsi que la plupart des autres troubles qui surviennent in utéro.
Résumé de l’auteur
De nombreux troubles cérébraux apparaissent in utero, y compris les troubles du spectre autistique (TSA). Ils sont générés par de nombreux événements pathogènes intrinsèques et extrinsèques, y compris des protéines non fonctionnelles produites en raison de mutation génétique, infections virales ou microbiennes maternelles, microbiote, signaux épigénétiques et immunitaires/inflammatoires.Ici, nous examinons d’un œil critique les limites intrinsèques de l’approche génomique et des outils utilisés pour évaluer l’origine génétique des TSA et la sous-évaluation des risques environnementaux pendant la maternité. Ces observations remettent en question l’existence d’un lien direct entre une mutation génétique et les séquelles cliniques en raison des rôles pathogènes cruciaux de la cascade d’altérations produite in utero par la mutation. Les principaux processus de développement, notamment la prolifération, la migration et la formation des synapses dépendent des gènes et de l’activité sont modulés par un large éventail de signaux moléculaires et non moléculaires. Ces signaux sont également vulnérables aux agressions environnementales qui peuvent retarder/dévier ces processus, entraînant des ensembles neuronaux mal connectés/mal placés qui sont la cause finale directe des séquelle et des cibles thérapeutiques possibles utilisant des antagonistes spécifiques de ces modèles immatures. La réunion de toutes les ressources et techniques nécessitera un large éventail d’approches scientifiques, y compris l’épidémiologie, la biologie et la médecine, la physiologie et la biologie du développement.Un défi interdisciplinaire que nous devons relever si nous voulons progresser dans l’atténuation et la guérison des TSAs.
Introduction
Le développement technologique extraordinaire et rapide de nouveaux outils génétiques au tournant du troisième millénaire a marqué le début d’une nouvelle ère dans les sciences biologiques, propice à une meilleure compréhension et le traitement des maladies. Plusieurs projets visaient à définir un ensemble de marqueurs bialléliques, polymorphismes de nucléotides simples (SNP) capturant la plupart des variations génomiques communes pour réaliser des études d’association à l’échelle du génome (GWAS) qui sont aujourd’hui considérées comme le meilleur moyen de disséquerles maladies humaines. Les revues présentent régulièrement de manière isolée que les GWAS ont révélé des variations communes de l’ADN communes qui affectent les risques de maladies courantes [1].
Cependant, les héritabilités dérivées des études pangénomiques sont étonnamment faibles [2] parce que les principales hypothèses requises pour utiliser les études d’association pangénomique ne sont jamais satisfaites dans les études humaines : (i) la maladie doit être homogène, (ii) générée par de nombreux facteurs génétiques indépendants, (iii) chacun doté de faibles effets, et que (iv) l’héritabilité de la maladie ne peut être déterminée que par des facteurs génétiques indépendant et (iv) ne doit pas interagir avec l’environnement. Ces conditions ne sont jamais remplies dans les études humaines, ce qui jette un doute sérieux sur leur utilisation en médecine [3]. Par conséquent, un changement complet de paradigme est changement complet de paradigme est nécessaire pour mieux comprendre et traiter les troubles.
Nous passons en revue les preuves qui remettent en question la validité d’une approche purement génétique des troubles du spectre autistique (TSA) qui sont très hétérogènes et dont les diagnostics reposent sur des déficits de communication sociale et de comportements répétitifs, ainsi que de nombreuses altérations sensorielles, motrices, gastro-intestinales, immunologiques, troubles anxieux, déficience intellectuelle, troubles mitochondriaux et épilepsie, affectant considérablement la santé, l’éducation, les soins sociaux, le bien-être, etc. [4]. Une telle hétérogénéité exclut l’hypothèse d’une simple relation génotype-phénotype utilisée pour l’estimation de l’héritabilité ce qui explique pourquoi les informations génomiques actuellement disponibles restent trop limitées pour permettre l’identification, et encore moins pour atténuer les déficiences des TSA dans la pratique clinique [examiné dans 5].
Études génétiques et études de jumeaux sur les TSA
Malgré d’intenses recherches, l’interprétation génétique de la plupart des formes de TSA reste discutable. Par exemple,une méta-analyse GWAS à grande échelle portant sur 18 381 patients atteints de TSA et 27 969 témoins a identifié de manière décevante seulement cinq loci significatifs à l’échelle du génome [6]. Même en ajoutant sept SNP détectés dans une étude d’association pangénomique multi-traits ne suffit pas à expliquer la totalité de l’héritabilité.Par conséquent, les estimations de l’héritabilité à l’aide de ces techniques, qui vont de 17 % à 91 % [7, revu dans 8], sont très controversées,car aucune des hypothèses des modèles statistiques n’est respectée dans ces analyses. En outre, 80,3 % des 85 des patients (n=218) atteints de TSA testés dans le cadre d’une autre étude ne présentaient aucune mutation génétique identifiable et des variantes positives de novo ont été observées chez 6,9 % des patients et dépendaient de l’âge des parents [9]. La comparaison de jumeaux monozygotes (MZ) et dizygotes (DZ) a été présentée comme une approche alternative et complémentaire pour déterminer l’hérédité des TSA souffre également de limitations intrinsèques. Les Jumeaux (MZ) sont le résultat d’un seul ovule fécondé, alors que les jumeaux DZ sont le résultat de deux ovules fécondés chacun par des spermatozoïdes différent fécondés par des spermatozoïdes différents. Une discordance pour un trait chez les jumeaux MZ est interprétée comme résultant de facteurs environnementaux différents, alors qu’un taux de concordance plus élevé chez les jumeaux MZ que chez les jumeaux DZ est interprété comme résultant des gènes, en ignorant les facteurs environnementaux [10]. Ces méthodes reposent sur deux hypothèses majeures non satisfaites, (i) les jumeaux MZ sont génétiquement identiques, alors que les jumeaux DZ partagent 50 % de leur génome, et (ii) l’exposition à l’environnement est équivalente pour les deux pendant tout le développement embryonnaire.L’hypothèse (i) selon laquelle les jumeaux MZ partagent la même composition génétique est toutefois remise en question par le fait que 15 % des mutations de novo sont spécifiques à l’un des jumeaux [11]. En outre, les jumeaux MZ partagent également le même placenta la plupart du temps (environ 70 %), ainsi que des amis, des classes, des soins parentaux, etc. dans une plus grande mesure que les jumeaux DZ ce qui ne peut qu’accroître leur ressemblance au-delà de leur similarité génétique [3]. Ces hypothèses ignorent donc l’impact des facteurs environnementaux [12], qui est souvent nié,et conduisent à des héritabilités surestimées. Les nombreux effets de méthylation de l’ADN médiés par le placenta [13] devraient rendre les épigénomes beaucoup plus similaires chez les jumeaux MZ que chez les jumeaux DZ [14], et en effet les génomes MZ sont identiques au début de la vie mais deviennent différents au fur et à mesure qu’ils grandissent [15].Le grand nombre de mutations génétiques rares identifiées, la plupart des enfants atteints de TSA ne bénéficient pas d’un diagnostic génétique spécifique, ce qui remet en question leur pertinence clinique et suggère une héritabilité génétique modérée avec un rôle substantiel des composantes environnementales partagées dans les TSA [16].
L’impact des mutations de novo
Les mutations de novo constituent le facteur génétique prédominant identifié dans les TSA [17]avec des mécanismes complexes encore mal compris. Dans les TSA non familiaux, les mutations ponctuelles de novo sont principalement d’origine paternelle et sont positivement corrélées à l’âge des parents [18]ce qui correspond à l’augmentation modeste mais significative du risque avec l’âge des parents [19]. Le fait que ces mutations de novo concernent de nombreux gènes (>500 gènes) remet en question l’idée d’un lien direct génotype-phénotype. Chacune de ces mutations affecte une multitude de gènes impliqués dans des voies fonctionnelles de base liées au développement des synapses, au ciblage des axones et à la motilité neuronal ce qui illustre le large éventail d’effets sur le développement des TSA [21].Seule une minorité de mutations de novo sont postzygotes. Générées par une seule ou plusieurs lignées cellulaires, les mutations pré-jumelage peuvent accroître les différences entre les génomes germinaux de jumeaux MZ. Une vaste étude sur l’influence parentale sur la lignée germinale en Islande décrit un niveau de diversité de séquence lié à des interactions complexes entre l’âge, le sexe, le type de mutation et la localisation génomique, [9, 22]ce qui implique une grande variété de facteurs d’interaction qui ne peuvent pas être facilement inclus dans un seul modèle génétique [23]. En outre, l’héritabilité ne peut être interprétée comme révélant la proportion de partage de l’ADN lorsque les variantes causales sont des mutations de novo ou impliquent des variants épigénétiques(voir section suivante). La nécessité d’identifier de nombreux biomarqueurs pour intégrer pleinement les multiples phénotypes associés aux TSA contraste avec le succès limité de l’identification des effets phénotypiques des variantes génétiques des TSA[24]. Il est désormais évident que la recherche sur l’autisme est coincée, comme l’illustrent les titres de revues pertinentes telles que « Searching for ways out of the autism maze : genetic, epigenetic and environmental » [25] ou »Lost in translation : traversing the complex path of genomics to therapeutics in autism spectrum Disorders »[26].En revanche, les affirmations selon lesquelles »les TSA ont une forte prédominance génétique [27] ou “ La conclusion que les TSA sont en grande partie génétique [19] ” est discutable.
L’hérédité des TSA et la transmission par des séquences autres que l’ADN
Des observations approfondies remettent en question l’hypothèse tacite selon laquelle il existe des liens directs entre les mutations et les séquelles cliniques et toutes les méthodes d’estimation de l’héritabilité et du risque supposent que l’hérédité résulte exclusivement de la transmission de la séquence d’ADN permettant ainsi de classer une maladie comme « génétique ». Cependant, les trois dernières décennies ont permis d’accumuler une quantité impressionnante d’informations bien documentés d’exemples bien documentés d’héritages non génétique chez la levure,les plantes, les invertébrés et les vertébrés, les mammifères et les humains,en lien notamment avec la pollution, concernant les troubles métaboliques (par exemple, troubles liés à l’alimentation comme le diabète), des troubles cardiaques, des testicules et des reins, et de nombreux traits comportementaux (dépression, stress mental, anxiété, peur, dépendance, addictions) [29,30].Les principaux canaux de transmission non génétiques sont l’épigénétique et le microbiote, ainsi que l’héritage culturel et écologique. Ceci est attesté par une pléthore d’études sur le sujet [31,32,33,34]. Un mécanisme générique d’action des effets environnementaux sur l’épigénome des cellules des cellules somatiques et germinales explique comment les facteurs environnementaux peuvent altérer l’intégrité de la lignée germinale, et donc le phénotype de la progéniture [35].Dans ce contexte, l’autisme représente un exemple classique de cas où l’hypothèse selon laquelle l’héritabilité signifie indubitablement la séquence de l’ADN peut piéger des générations de chercheurs.La course aux gènes impliqués (tableau complémentaire en ligne) n’a conduit à aucune percée conceptuelle ou thérapeutique. La propriété dynamique intrinsèque des réseaux cérébraux développementaux et la plasticité réactive en réponse à une pathologie génétique ou environnementale dissocient effectivement les phénotypes des TSA de tout événement pathologique génétique inaugural. L’identification d’une architecture génétique synaptique de l’ASD reste purement corrélationnelle, remettant en question la causalité génotype-phénotype et ne suggère pas de cibles thérapeutiques spécifiques. Il est intéressant de noter que le nombre de gènes impliqués dans les TSA augmente de façon exponentielle en relation avec l’amélioration des méthodes de tests génétiques [27] illustrant la difficulté à relier directment généotype et phénotype.
La contribution de l’axe intestin-cerveau-immunité
Dans le cas de troubles tels que les TSA, qui se manifestent dès la naissance, de nombreuses agressions environnementales extrinsèques et intrinsèques altèrent le développement normal de l’enfant. Parmi ces nombreuses agressions in utero environnementales, les interactions intestin-cerveau-immunitaires ont été directement liées aux TSA, y compris à la pathogenèse, et sont souvent sous-estimées (tableau 1, figure 1 et travaux antérieurs [8,36]. En 1998, reconnaissant que (1) la plupart des enfants autistes présentent un dysfonctionnement douloureux de l’intestin, [5,8] (2) beaucoup d’entre eux ont des antécédents de traitement antibiotiques au début de leur vie et (3) que de nombreux antibiotiques n’affectent pas les Clostridia, une classe de bactéries qui se trouvent dans l’intestin, une classe de bactéries connues pour produire des métabolites neurotoxiques; Bolte a suggéré que le microbiote intestinal pourrait être impliqué dans l’autisme. Elle a testé cette hypothèse non conventionnelle en traitant son propre enfant autiste avec de la vancomycine, un antibiotique qui affecte les Clostridies et a constaté que cela améliorait radicalement le comportement de son fils, ce qui l’a amenée, avec ses collègues, à tester sur un panel plus large, ce qui a confirmé son intuition [38].Depuis,le séquençage à haut débit a considérablement amélioré notre capacité d’étudier notre microbiote, ce qui a permis de démontrer l’implication du microbiote intestinal dans les TSA (voir tableau 1 ). Par exemple, il a été que le microbiote intestinal a un impact sur le développement du cerveau des mammifères et sur leur comportement à l’âge adulte[39] . De plus, l’injection de microbiote d’humains autistes sur des souris sans germes a conduit à des comportements autistiques caractéristiques [38]. En outre, deux articles portant sur le même échantillon de 18 patients ont montré que le transfert de microbiote d’individus neurotypiques à des patients autistes a conduit à de fortes améliorations qui ont persisté pendant 8 semaines après le transfer et, deux ans plus tard, tous les symptômes et comportements s’amélioraient encore [41].Cette étude ouvre non seulement la porte à la possibilité de traiter à long terme les patients atteints de TSA, mais elle montre également une fois pour toutes que le microbiote joue un rôle prépondérant dans les TSA. Il est concevable que l’héritabilité des TSA en partie résulte d’un transfert de la mère à l’enfant du microbiote intestinal à la naissance et par le colostrum, [42] et de l’action sélective forte du régime alimentaire sur le microbiote [43], le régime alimentaire étant probablement lui-même héréditaire par transmission culturelle.

En outre, l’inflammation maternelle mérite une attention particulière dans la pathogenèse des TSA. La neuroinflammation est provoquée par des insultes virales ou microbiennes, des insultes immunitaires, la fièvre, les troubles cardiovasculaires, l’obésité, les maladies auto-immunes, et le stress produit par la séparation d’avec la mère juste après la naissance [44,47]. L’activation immunitaire maternelle constitue un lien de causalité menant à des dysfonctionnements neuronaux et à des phénotypes comportementaux des années ou des décennies plus tard chez l’adulte ou l’enfant [48]. L’infection prénatale de rongeurs gravides par le virus de la grippe génère à long terme des changements fonctionnels,comportementaux et structurelles du cerveau chez la progéniture, comme celles observées chez les patients [49].L’utilisation maternelle du valproate de sodium, provoque également des TSA chez les modèles animaux et chez l’homme [50,51]. Les pesticides prénataux augmentent l’incidence des accouchements prématurés [52]et l’incidence des TSA [53,54].La consommation de drogues légales ou illégales pendant la grossesse affecte également le développement du cerveau induisant des maladies cérébrales [55]. Le lien avec le mécanisme sous-jacent comprend l’activation immunitaire précoce, les signaux inflammatoires impliquant les interleukines et d’autres signaux qui font dérailler des processus de développement essentiels y compris la prolifération cellulaire et la migration neuronale. Il est important de noter que les événements génétiques et environnementaux peuvent interagir de manière séquentielle, pour produire les effets délétères, ce qui illustre la complexité de la maladie, mais aussi remettent en question toute approche reposant uniquement sur les mutations génétiques et qui postule l’existence d’un lien direct entre la mutation et la cause ultime des séquelles.
Identification précoce des TSA
Deux études complémentaires ont montré que des altérations de développement in utero modifient de manière significative les futurs bébés atteints de TSA, au point qu’ils peuvent être identifiés avec une précision raisonnable dès la naissance. Dans le premier cas, la comparaison de la croissance du cerveau à la fin de la gestation entre les fœtus qui développeront ou non plus tard un TSA a montré une croissance cérébrale excessive à partir de la seconde moitié du deuxième trimestre de la gestation (22e semaine d’aménorrhée) [56]. La seconde étude, plus détaillée, a utilisé plus de 140 paramètres non génétiques recueillis dans les maternités françaises auprès de 63 bébés atteints de TSA et de 189 bébés neurotypiques nés au cours de la même année, dans la même maternité et dans les mêmes conditions [57].Un programme d’apprentissage automatique (Machine Learning) a permis d’identifier à la naissance près de la moitié des futurs bébés atteints de TSA avec un haut degré de spécificité et pratiquement tous les bébés neurotypiques (96 %). Après validation dans de nombreuses maternités, un test pourrait être mis au point et appliqué afin d’identifier dès la naissance les bébés qui seront plus tard diagnostiqués avec un TSA. Les mesures pertinentes comprennent les mesures attendues (infections virales ou microbiennes), mais aussi des paramètres inattendus tels que une plus grande taille du fémur au cours du deuxième trimestre, ou une position plus précoce face vers le bas avant la naissance. En outre, la circonférence de la tête du fœtus (HC) est significativement plus grand au cours du troisième trimestre de gestation chez 38 % des fœtus qui développeront un TSA, par rapport aux fœtus neurotypiques appariés à l’âge (figure 2).Cela correspond à la plus grande circonférence cérébrale observée chez environ 40 % des bébés et des nourrissons atteints de TSA [58].Il est intéressant de noter que la troisième échographie réalisée peu avant l’accouchement montre que tous les « futurs TSA » ont une plus grande circonférence cérébrale que celui des fœtus neurotypiques correspondants ce qui suggère une déficience d’un processus essentiel de préparation à la naissance. En outre, l’HC d’environ 40 % des futurs enfants atteints de TSA est plus grand que celui des autres fœtus déjà au deuxième trimestre (figure 2, panneau de droite), ce qui suggère que l’HC plus importante rapportée chez les enfants et les adolescents est visible in utero. Cette hypothèse a été validée expérimentalement chez les rongeurs, où le volume de l’hippocampe et du néocortex est plus important après la naissance que peu avant chez les « souris TSA » [59].Plus surprenant peut-être, la comparaison de la taille des neurones de l’hippocampe peu avant et après l’accouchement révèle que les neurones Les neurones et le volume de la structure augmentent donc pendant la naissance, ce qui n’est pas le cas chez les controles. Clairement, les TSAs sont nés in utéro et pendant la naissance, avec des modifications structurales précoces.

Ceci est attesté par des signatures anatomo-pathologiques telles que des plaques de malformations corticales et des colonnes corticales altérées, et des modifications du nombre de neurones et de la taille des structures cérébrales [60].Il est donc nécessaire d’établir des méthodes de diagnostic fiables pour détecter les enfants autistes dès la naissance. Pourtant, dans la plupart des pays développés,malgré les recommandations des académies de médecine et des autorités politiques, le diagnostic n’est établi que vers 4-5 ans.La détection tardive laisse passer la meilleure fenêtre d’opportunité pour le traitement et conduit à une perte de la meilleure opportunité de traitement et entraîne un handicap à vie qui aurait pu être considérablement atténué par un traitement précoce (figure 3). L’importance de la détection et de l’intervention précoceset l’intervention précoces pour guérir les enfants atteints de TSA ont été développés ailleurs [61].

Périodes critiques et temporalité des troubles du développement
Dans le développement du cerveau, « la seule constante est le changement » (Ephesus,c. 535-475 AV. J.-C.). Le cerveau en développement subit d’importantes modifications. Plusieurs étapes essentielles, notamment la prolifération cellulaire, la migration, la formation de synapses et d’ensembles neuronaux, sont en grande partie propres au cerveau en développement. Pratiquement tous les courants ioniques, y compris les courants portés par le voltage ou les transmetteurs, et les activités des réseaux de neurones diffèrent fortement et exercent des fonctions différentes dans le cerveau en développement et le cerveau adulte [62-65]. En résumé, le cerveau en développement n’est pas un petit cerveau adulte, mais un cerveau doté de fonctions très spécifiques qui doivent se dérouler en temps voulu.En outre, le développement du cerveau n’est pas un processus linéaire continu, car il comporte de nombreuses « périodes critiques » représentant des étapes dans l’établissement d’entités fonctionnelles. L’altération de ces périodes critiques entraîne de graves séquelles chez l’homme et l’animal [66].Par exemple, les troubles de la migration produits par des mutations génétiques ou des agressions environnementales et inflammatoires pendant la période de migration corticale sont l’une des principales causes d’épilepsies pharmacorésistantes et de la déficience intellectuelle. [60-67]. De même, des altérations qui se produisent pendant la période période critique du système visuel ont un impact sur son développement [68].Chez le fœtus prématuré, les mouvements spontanés sont associés à une activité électrique qui se propage de la périphérie vers le centre, agissant comme un système de rétroaction sensorielle qui contrôle la formation des cartes corticales [69].Les traitements médicamenteux ou les manipulations comportementales comme les interventions pharmacologiques peuvent avancer ou retarder ces périodes critiques avec des conséquences à long terme [70].
La parturition et la naissance sont également des périodes critiques majeures. Le stress de la naissance » est associé à une libération massive d’hormones de stress nécessaires aux fonctions respiratoires[71]mais potentiellement toxiques pour l’activité neuronale. L’ocytocine, qui déclenche l’accouchement, atténue également les effets des hormones de stress exerçant de plus des actions analgésiques[72-73]. Il est intéressant de noter que l’HC fœtale juste avant la naissance est plus importante chez les futurs enfants atteints de TSA que chez les enfants neurotypiques appariés selon l’âge, ce qui suggère qu’un événement protecteur avant la naissance a été aboli dans les TSA (figure 2). Dans le même ordre d’idées, dans les modèles de rongeurs, le volume de l’hippocampe et des neurones hippocampiques augmente à la naissance chez les « souris TSA », mais pas chez les contrôle.
La prématurité et, dans une moindre mesure, la césarienne sont associés à une augmentation de l’incidence des TSA [74-75[62-65]. les mécanismes sous jacents et signaux inflammatoires in utero, doivent être pris en compte dans les modèles visant à expliquer la pathogenèse des TSA.
Lost in translation : l’approche purement génomique est une une impasse thérapeutique
L’objectif premier de l’application des outils génomiques aux TSA était de de mieux comprendre les processus pathogènes et de suggérer de nouvelles pistes thérapeutiques pour les TSA. Malgré des efforts considérables. Ces objectifs n’ont pas été atteints , même dans des syndromes bien caractérisées comme le X fragile (FRX). En effet, en tant que maladie monogénique, on pensait que le FRX aurait des cibles médicamenteuses cohérentes qui pourraient être modulées par divers médicaments. Cependant, malgré des observations expérimentales prometteuses, la traduction thérapeutique de ces donnéess a largement échoué. [77]. Les raisons de ces échecs sont l’hétérogénéité clinique du FRX, qui nécessite l’identification de sous-populations répondant au traitement [78]. Les mutations uniques associées au FRX (ou au syndrome de Rett)sont couramment associées aux TSA et ont des cibles complexes avec de multiples cascades d’altérations déjà in utero, ce qui suggère de multiples séries d’altérations précoces et empêchant d’envisager une relation directe entre une seule mutation et un syndrome. Toutefois, plus récemment, un essai préliminaire utilisant un inhibiteur de la phosphodiestérase (BPN 14770) a montré des effets prometteurs sur des enfants atteints de FRX, [79] bien que l’hétérogénéité du FRX va nécessiter es essais de plus grande envergure, comme dans le cas des TSA.
Il est important de souligner les nombreux changements importants qui se produisent au cours du développement du cerveau.Les schémas de fonctionnement diffèrent dans les cerveaux immatures et adultes et exercent des rôles différents. Dans le cerveau en développement, l’activité va moduler la prolifération neuronale, la migration, la formation des synapses et et même le phénotype cellulaire : dans la moelle épinière des amphibiens, le phénotype GABA ou glutamate est modulé par l’activité neuronale, le phénotype GABA ou glutamate est déterminé par l’activité en cours. Le concept de « check point » postule que l’activité du réseau en développement et l’expression des gènes constituent des points de passage obligés qui contrôlent la synchronisation et l’activation des processus développementaux essentiels [80] . La modification ou l’impact de cette séquence a des effets de longue durée et séquelles importantes et durables. Ainsi, l’augmentation de la fréquence des courants calciques dans les neurones glutamatergiques excitateurs change leur phénotype les transformant en neuones inhibiteurs (ligérant du GABA) et inversement pour les neurones GABAergiques, préservant ainsi l’équilibre excitateur/inhibiteur (figure 4, panneau de gauche) [81]. Un changement au cours du développement parmi les mieux décrits, est le shift des actions du GABA qui excite les neurones immatures et inhibe les neurones adultes. Ce shift a été préservé tout au long de l’évolution. Le GABA exerce un large éventail d’actions trophiques, notamment sur la croissance cellulaire, les synapses et des réseaux [63].Ce changement de développement est dû à une baisse progressive des niveaux de (Cl-)i qui sont élevés dans les jeunes neurones et faibles chez les adultes (figure 4, panneau de droite). Cette altération développementale est due à l’activité accrue de du co-transporteurs d’ions – NKCC1- dans les neurones immatures se traduisant par des concentrations plus élevés de chlore dans les cellules immatures [63,82].Ces propriétés se traduisent par desactivités électriques qui sont uniques au cerveau en développement chez les rongeurs et les primates primates non humainsavec des des schémas équivalents dans l’activité humaine [69,85]. Il est intéressant de noter que des événements pathologiques survenant au cours de ces points de contrôle, entraînent des altérations permanentes, avec notamment des ensembles neuronaux mal placés ou mal connectés. Dans le même ordre d’idées, dans un large éventail de troubles, y compris les TSA, diverses épilepsies, l’X fragile, le syndrome de Down, etc, on observe une activité exacerbée de NKCC1, avec des neurones restés dotés de propriétés immatures. Le concept de « neuroarchéologie » postule que ces altérations persistantes sont la cause finale des troubles nés in utero et des cibles thérapeutiques possibles en utilisant des inhibiteurs sélectifs del’activité générée par ces neurones immatures [86].
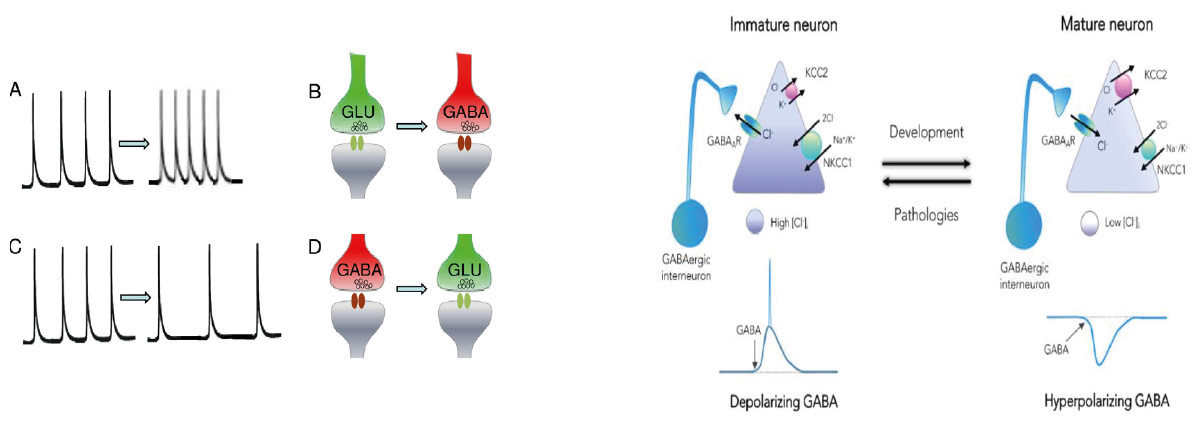
Plusieurs essais ont testé les effets d’un inhibiteur sélectif de de NKCC1 pour atténuer les es actions excitatrices du GABA. L’inhibiteur de l’importateur de chlorure l’importateur de NKCC1, le bumétanide, a permis de restaurer l’inhibition GABAergique dans des modèles de TSA dans divers modèles animaux pathologiques. Un essai monocentrique [87] suivie d’une phase II multicentrique [88]en double aveugle,ont montré une différence statistiquement significative entre les enfants traitrés par la bumétanide et le placebo. En utilisant les conditions et à la même dose, des essais de phase II similaires ont été menés avec succès en Chine (trois essais), aux Pays-Bas, en Iran, Égypte et en Tunisie, avec un total de plus de 1036 enfants traités selon une méta-analyse [89]. Les mesures de suivi oculaire et l’imagerie cérébrale ont montré respectivement, une amélioration significative du contact visuel et une réduction de l’activation de l’amygdale dans des contacts visuels que les autistes souvent évitent [90,91].Malheureusement, deux derniers essais de phase III réalisés dans de nombreux pays européens, au Brésil et en Australie (210 enfants âgés de 2 à 6 ans et 210 enfants âgés de 7 ans)n’ont pas permis d’observer une différence significative entre les enfants traités et placébo [92]. Ceci est conforme à l’échec répété des grands essais de phase III de nombreux troubles neurodéveloppementaux, malgré des essais de phase II prometteurs. Cela est très probablement dû à l’hétérogénéité des TSA et à la nécessité d’identifier des sous-populations et de mieux stratifier les critères d’inclusion en en fonction de marqueurs cliniques ou biologiques. Cela suggère qu’il est peu probable qu’un traitement unique pour tous les phénotypes de TSA soit probable. En d’autres termes, le développement futur des traitements des TSA est conditionné par l’identification clinique de phénotypes homogènes d’enfants atteints de TSA à l’aide de l’IA. Le concept de l’« origine fœtale des troubles » a été développé aussi pour le diabète et les maladies cardiovasculaire[94].
CONCLUSIONS ET PERSPECTIVES D’AVENIR
Notre analyse nous amène à proposer une stratégie thérapeutique (figure 3).Les TSA, comme beaucoup d’autres troubles neurodéveloppementaux, sont très hétérogène ce qui « tempère l’espoir de découvertes étiologiques capitales dans un avenir proche, qui permettraient d’identifier des traitements spécifiques, mécanistes et curatifs pour ces conditions » [61]. Loin d’être un primus inter pares, l’information génétique n’est que l’un des nombreux facteurs qui régissent le développement du cerveau, et par conséquent, le traitement des TSA et d’autres troubles neurodéveloppementaux est conditionné par une meilleure compréhension de leur pathogenèse développementale. Les troubles nés in utero, tels que les TSA,nécessitent un changement méthodologique complet, passant d’une approche purement génomique à une approche qui inclut un large éventail de facteurs et d’expertises. L’étape clé de ce changement consiste à détecter les troubles le plus tôt possible afin d’utiliser la période cruciale du développement caractérisée par une plasticité exceptionnelle au cours des années qui suivent la naissance pour au moins atténuer la plupart des déficiences des TSA (figure 3). Par exemple, l’avantage d’un diagnostic précoce a été démontré dans le cas de problèmes d’audition ou de vision [95].Malheureusement, plus de 50 % des patients ne sont identifiés qu’à l’école en raison de leur comportement gravement perturbateur et de leurs faibles capacités sociales et de communication .L’identification précoce des TSA implique également de redéfinir la séquence des étapes et des points de contrôle du développement précoce du cerveau. Nous postulons que l’hétérogénéité des TSA est le sous-produit du stade maturatif correspondant aux évènements pathologiques et traumatisme génétique-environnemental initial. Ainsi, l’identification et la validation des biomarqueurs des TSA (tels que l’HC) doivent conduire à la collecte de données sur le développement dans les maternités et devraient faire partie intégrante du programme de recherche et devraient faire partie des protocoles de suivi de routine des femmes enceintes pour permettre leur généralisation pour détecter et traiter les futurs patients atteints de TSA dès la naissance et d’utiliser des techniques psycho-éducatives pour renforcer les interactions sociales. Certes, cette tâche implique de modifier les protocoles de suivi des femmes enceintes dans plusieurs pays ou elle n’est pas aussi développée qu’en France, mais cela ne devrait pas être une difficulté majeure au vu des bénéfices importants potentiels. En outre, la collecte massive de ce type de données permettra à son tour d’améliorer notre compréhension de l’hétérogénéité des TSA. Un tel protocole de routine peut également être complété par la collecte de données épigénétiques dans des sous-échantillons spécifiques, car l’épigénétique semble clairement jouer un rôle important dans les TSA. Il est désormais essentiel de remplacer l’idée que les TSA sont une maladie « génétique » par l’idée plus précise que les TSA sont un « syndrome multifactoriel » avec un possible risque génétique ». Cela nous permettra également de passer de l’impossible « guérison complète de la maladie » à « optimiser les résultats » afin de mieux comprendre et atténuer les séquelles délétères. En fait, l’objectif d’un traitement serait plutôt de s’assurer que les déficiences innées ne se traduisent pas par des handicaps permanents [61].Ce changement conceptuel reflétera également la prise de conscience du fait que les TSA et d’autres troubles neurodéveloppementaux sont des « extrémités de dimensions distribuées de façon continue, un peu comme l’hypertension sur le continuum de la pression sanguine » [ou de la pression artérielle » [61]. Dans une perspective thérapeutique, le concept de neuroarchéologie devrait permettre d’identifier des activités immatures persistantes exprimées dans des conditions expérimentales qui peuvent être blouqées par des médicaments sélectifs. Le changement excitateur de GABA avec l’inhibiteur NKCC1 n’est qu’un exemple de cette de cette approche. L’identification des réseaux aberrants générés par l’insulte inaugurale devrait aider à développer des médicaments qui réduiraient spécifiquement au silence ces réseaux « immatures ». En outre, il convient de souligner que, nos conclusions sont valables pour de nombreux autres troubles neurodéveloppementaux, dont certains d’origine génétique associés à des symptômes de type TSA chez lesquels, on observe aussi une activité NKCC1 accrue et des effets excitateurs du GABA et notamment, l’X Fragile, La trisomie 21, le syndrome de Rett,ou celui du dundrome de la délétion 22q-11-2 [85, 96-100]. Pour chacun d’entre eux, des thérapeutiques potentielles similaires ont été mises au point utilisant la bumétanide et d’autres stratégies visant à réduire l’activité de la NKCC1ont également été suggérées.Par conséquent, nos conclusions ont une portée beaucoup plus large que la seule application aux TSA, qui sert ici d’exemple de la nécessité générale d’intégrer toutes les formes de toutes les formes d’hérédité dans nos approches médicales.
En conclusion, nous demandons instamment aux chercheurs et aux praticiens à dépasser la vision déterministe réductrice de la vie qui se concentre trop sur la séquence de l’ADN et d’adopter une approche qui intègre les informations provenant de tous les domaines de la biologie.Cette approche devrait prendre en compte la temporalité de l’apparition des troubles et la propriété dynamique fondamentale du cerveau afin de pouvoir détecter et agir avant que des dommages irréversibles ne soient causés. Pour réunir toutes les ressources et les techniques, il faudra s’appuyer sur de nombreuses approches scientifiques, dont l’épidémiologie, l’anatomie, la pathologie, la microbiologie, la physiologie et la biologie du développement, un défi interdisciplinaire que nous devons relever si nous voulons progresser dans la lutte contre de nombreuses maladies complexes.
SOURCES
1 Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 2017;101:5–22.
2 Maher B. Personal genomes: The case of the missing heritability. Nature New Biol 2008;456:18–21.
3 Robette N, Génin E, Clerget-Darpoux F. Heritability: What’s the point? What is it not for? A human genetics perspective. Genetica 2022;150:199–208.
4 Hodges H, Fealko C, Soares N. Autism spectrum disorder: definition, epidemiology, causes, and clinical evaluation. Transl Pediatr 2020;9:S55–65.
5 Puricelli C, Rolla R, Gigliotti L, et al. The Gut-Brain- Immune Axis in Autism Spectrum Disorders: A State-of- Art Report. Front Psychiatry 2021;12:755171.
6 Grove J, Ripke S, Als TD, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019;51:431–44.
7 Bourgeron T. From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci 2015;16:551–63.
8 Nitschke A, Deonandan R, Konkle ATM. The link between autism spectrum disorder and gut microbiota: A scoping review. Autism 2020;24:1328–44.
9 Geier DA, Kern JK, Sykes LK, et al. Examining genotypic variation in autism spectrum disorder and its relationship to parental age and phenotype. Appl Clin Genet 2016;9:121–9.
10 Falconer DS. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genet 1965;29:51–76.
11 Jonsson H, Magnusdottir E, Eggertsson HP, et al. Differences between germline genomes of monozygotic twins. Nat Genet 2021;53:27–34.
12 Bölte S, Girdler S, Marschik PB. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci 2019;76:1275–97.
13 Bahado-Singh RO, Vishweswaraiah S, Aydas B, et al. Artificial intelligence and placental DNA methylation: newborn prediction and molecular mechanisms of autism in preterm children. J Matern Fetal Neonatal Med 2025;35:8150–9.
14 van Dongen J, Gordon SD, McRae AF, et al. Identical twins carry a persistent epigenetic signature of early genome programming. Nat Commun 2021;12:5618.
15 Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 2005;102:10604–9.
16 Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011;68:1095–102.
17 Weiner DJ, Wigdor EM, Ripke S, et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 2017;49:978–85.
18 Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature New Biol 2012;488:471–5.
19 Sandin S, Yip BHK, Yin W, et al. Examining Sex Differences in Autism Heritability. JAMA Psychiatry 2024;81:673.
20 O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature New Biol 2012;485:246–50.
21 Gilman SR, Iossifov I, Levy D, et al. Rare De Novo Variants Associated with Autism Implicate a Large Functional Network of Genes Involved in Formation and Function of Synapses. Neuron 2011;70:898–907.
22 Jónsson H, Sulem P, Kehr B, et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature New Biol 2017;549:519–22.
23 Wroten M, Yoon S, Andrews P, et al. Sharing parental genomes by siblings concordant or discordant for autism. Cell Genom 2023;3:100319.
24 Rolland T, Cliquet F, Anney RJL, et al. Phenotypic effects of genetic variants associated with autism. Nat Med 2023;29:1671–80.
25 Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci 2006;29:349–58.
26 Sestan N, State MW. Lost in Translation: Traversing the Complex Path from Genomics to Therapeutics in Autism Spectrum Disorder. Neuron 2018 ;100:406–23.
27 Kereszturi É. Diversity and Classification of Genetic Variations in Autism Spectrum Disorder. Int J Mol Sci 2023;24:16768.
28 Perez MF, Lehner B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat Cell Biol 2019;21:143–51.
29 Zeid D, Gould TJ. Impact of nicotine, alcohol, and cocaine exposure on germline integrity and epigenome. Neuropharmacology 2020;173.
30 Bohacek J, Mansuy IM. Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet 2015;16:641–52.
31 Wang Y, Liu H, Sun Z. Lamarck rises from his grave: parental environment-induced epigenetic inheritance in model organisms and humans. Biol Rev Camb Philos Soc 2017;92:2084–111.
32 Danchin E, Pocheville A, Rey O, et al. Epigenetically facilitated mutational assimilation: epigenetics as a hub within the inclusive evolutionary synthesis. Biological Rev 2019;94:259–82.
33 Fitz-James MH, Cavalli G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat Rev Genet 2022;23:325–41.
34 Danchin E. From the modern synthesis to the inclusive evolutionary synthesis: an einsteinian revolution in evolution. In: du Crest A, Valković M, Ariew A, et al, eds. Evolutionary thinking across disciplines; problems and perspectives in generalized Darwinism. Switzerland: Springer Nature Switzerland AG, 2023: 401–27.
35 Danchin É, Pocheville A, Huneman P. Early in life effects and heredity: reconciling neo-Darwinism with neo-Lamarckism under the banner of the inclusive evolutionary synthesis. Phil Trans R Soc B 2019;374:20180113.
36 Cryan JF, O’Riordan KJ, Cowan CSM, et al. The Microbiota-Gut- Brain Axis. Physiol Rev 2019;99:1877–2013.
37 Bolte ER. Autism and Clostridium tetani. Med Hypotheses 1998;51:133–44.
38 Sandler RH, Finegold SM, Bolte ER, et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J Child Neurol 2000;15:429–35.
39 Heijtz RD, Wang S, Anuar F, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 2011;108:3047–52.
40 Sharon G, Cruz NJ, Kang D-W, et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019;177:1600–18.
41 Kang D-W, Adams JB, Vargason T, et al. Distinct Fecal and Plasma Metabolites in Children with Autism Spectrum Disorders and Their Modulation after Microbiota Transfer Therapy. mSphere 2020;5:e00314-20.
42 Toquet M, Gómez-Martín Á, Bataller E. Review of the bacterial composition of healthy milk, mastitis milk and colostrum in small ruminants. Res Vet Sci 2021;140:1–5.
43 Salonen A, de Vos WM. Impact of diet on human intestinal microbiota and health. Annu Rev Food Sci Technol 2014;5:239–62.
44 Edlow AG. Maternal obesity and neurodevelopmental and psychiatric disorders in offspring. Prenat Diagn 2017;37:95–110.
45 Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci 2017;20:145–55.
46 Han VX, Patel S, Jones HF, et al. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat Rev Neurol 2021;17:564–79.
47 Modabbernia A, Velthorst E, Reichenberg A. Environmental risk factors for autism: an evidence-based review of systematic reviews and meta-analyses. Mol Autism 2017;8:13.
48 Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013;155:1451–63.
49 Shi L, Fatemi SH, Sidwell RW, et al. Maternal Influenza Infection Causes Marked Behavioral and Pharmacological Changes in the Offspring. J Neurosci 2003;23:297–302.
50 Christensen J, Grønborg TK, Sørensen MJ, et al. Prenatal Valproate Exposure and Risk of Autism Spectrum Disorders and Childhood Autism. JAMA 2013;309:1696.
51 Ingram JL, Peckham SM, Tisdale B, et al. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol Teratol 2000;22:319–24.
52 Kolan AS, Hall JM. Association of Preterm Birth and Exposure to Endocrine Disrupting Chemicals. Int J Mol Sci 2023;24:1952.
53 Shelton JF, Geraghty EM, Tancredi DJ, et al. Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides: the CHARGE study. Environ Health Perspect 2014;122:1103–9.
54 Mostafalou S, Abdollahi M. Pesticides: an update of human exposure and toxicity. Arch Toxicol 2017;91:549–99.
55 Thompson BL, Levitt P, Stanwood GD. Prenatal exposure to drugs: effects on brain development and implications for policy and education. Nat Rev Neurosci 2009;10:303–12.
56 Bonnet-Brilhault F, Rajerison TA, Paillet C, et al. Autism is a prenatal disorder: Evidence from late gestation brain overgrowth. Autism Res 2018;11:1635–42.
57 Caly H, Rabiei H, Coste-Mazeau P, et al. Machine learning analysis of pregnancy data enables early identification of a subpopulation of newborns with ASD. Sci Rep 2021;11:6877.
58 Libero LE, Nordahl CW, Li DD, et al. Persistence of megalencephaly in a subgroup of young boys with autism spectrum disorder. Autism Res 2016;9:1169–82.
59 Cloarec R, Riffault B, Dufour A, et al. Pyramidal neuron growth and increased hippocampal volume during labor and birth in autism. Sci Adv 2019;5:eaav0394.
60 Casanova MF, Buxhoeveden DP, Switala AE, et al. Minicolumnar pathology in autism. Neurology (ECronicon) 2002;58:428–32.
61 Klin A, Micheletti M, Klaiman C, et al. Affording autism an early brain development re-definition. Dev Psychopathol 2020;32:1175–89.
62 Spitzer NC, Borodinsky LN. Implications of activity-dependent neurotransmitter– receptor matching. Phil Trans R Soc B 2008;363:1393–9.
63 Ben-Ari Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 2002;3:728–39.
64 Chalupa LM. Retinal waves are unlikely to instruct the formation of eye-specific retinogeniculate projections. Neural Dev 2009;4:1–7.
65 Ben-Ari Y, Gaiarsa J-L, Tyzio R, et al. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 2007;87:1215–84.
66 Desai NS, Cudmore RH, Nelson SB, et al. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci 2002;5:783–9.
67 Ackman JB, Aniksztejn L, Crépel V, et al. Abnormal network activity in a targeted genetic model of human double cortex. J Neurosci 2009;29:313–27.
68 Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr Opin Neurobiol 2000;10:138–45.
69 Milh M, Kaminska A, Huon C, et al. Rapid cortical oscillations and early motor activity in premature human neonate. Cereb Cortex 2007;17:1582–94.
70 Marguet SL, Le-Schulte VTQ, Merseburg A, et al. Treatment during a vulnerable developmental period rescues a genetic epilepsy. Nat Med 2015;21:1436–44.
71 Lagercrantz H, Slotkin TA. The “stress” of being born. Sci Am 1986;254:100–7.
72 Tyzio R, Nardou R, Ferrari DC, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 2014;343:675–9.
73 Mazzuca M, Minlebaev M, Shakirzyanova A, et al. Newborn Analgesia Mediated by Oxytocin during Delivery. Front Cell Neurosci 2011;5:3.
74 Curran EA, O’Neill SM, Cryan JF, et al. Research review: Birth by caesarean section and development of autism spectrum disorder and attention-deficit/ hyperactivity disorder: a systematic review and meta-analysis. J Child Psychol Psychiatry 2015;56:500–8.
75 Padilla N, Eklöf E, Mårtensson GE, et al. Poor Brain Growth in Extremely Preterm Neonates Long Before the Onset of Autism Spectrum Disorder Symptoms. Cereb Cortex 2017;27:1245–52.
76 Myers SM, Challman TD, Bernier R, et al. Insufficient Evidence for “Autism-Specific” Genes. Am J Hum Genet 2020;106:587–95.
77 Erickson CA, Davenport MH, Schaefer TL, et al. Fragile X targeted pharmacotherapy: lessons learned and future directions. J Neurodev Disord 2017;9:7:7:.
78 Verdura E, Pérez-Cano L, Sabido-Vera R, et al. Heterogeneity in Fragile X Syndrome Highlights the Need for Precision Medicine-Based Treatments. Front Psychiatry 2021;12:722378.
79 Berry-Kravis EM, Harnett MD, Reines SA, et al. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: a randomized, placebo-controlled, phase 2 clinical trial. Nat Med 2021;27:862–70.
80 Ben-Ari Y, Spitzer NC. Phenotypic checkpoints regulate neuronal development. Trends Neurosci 2010;33:485–92.
81 Spitzer NC. Neurotransmitter Switching in the Developing and Adult Brain. Annu Rev Neurosci 2017;40:1–19.
82 Rivera C, Voipio J, Payne JA, et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature New Biol 1999;397:251–5.
83 Leinekugel X, Khazipov R, Cannon R, et al. Correlated bursts of activity in the neonatal hippocampus in vivo. Science 2002;296:2049–52.
84 Khazipov R, Esclapez M, Caillard O, et al. Early development of neuronal activity in the primate hippocampus in utero. J Neurosci 2001;21:9770–81.
85 He Q, Nomura T, Xu J, et al. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci 2014;34:446–50.
86 Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci 2008;31:626–36.
87 Lemonnier E, Degrez C, Phelep M, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry 2012;2:e202.
88 Lemonnier E, Villeneuve N, Sonie S, et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 2017;7:e1056.
89 Xiao H-L, Zhu H, Jing J-Q, et al. Can bumetanide be a miraculous medicine for autism spectrum disorder: Meta-analysis evidence from randomized controlled trials. Res Autism Spectr Disord 2024;114:102363.
90 Hadjikhani N, Åsberg Johnels J, Lassalle A, et al. Bumetanide for autism: more eye contact, less amygdala activation. Sci Rep 2018;8:3602.
91 Hadjikhani N, Zürcher NR, Rogier O, et al. Improving emotional face perception in autism with diuretic bumetanide: a proof-of- concept behavioral and functional brain imaging pilot study. Autism 2015;19:149–57.
92 Fuentes J, Parellada M, Georgoula C, et al. Bumetanide oral solution for the treatment of children and adolescents with autism spectrum disorder: Results from two randomized phase III studies. Autism Res 2023;16:2021–34.
93 Juarez-Martinez EL, van Andel DM, Sprengers JJ, et al. Bumetanide Effects on Resting-State EEG in Tuberous Sclerosis Complex in Relation to Clinical Outcome: An Open-Label Study. Front Neurosci 2022;16:879451.
94 Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 2014;94:1027–76.
95 Korver AMH, Smith RJH, Van Camp G, et al. Congenital hearing loss. Nat Rev Dis Primers 2017;3:16094.
96 LaSalle JM. Epigenomic signatures reveal mechanistic clues and predictive markers for autism spectrum disorder. Mol Psychiatry 2023;28:1890–901.
97 Deidda G, Parrini M, Naskar S, et al. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat Med 2015;21:318–26.
98 Ip JPK, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 2018;19:368–82.
99 Smrt RD, Eaves-Egenes J, Barkho BZ, et al. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis 2007;27:77–89.
100 Amin H, Marinaro F, De Pietri Tonelli D, et al. Developmental excitatory-to- inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci Rep 2017;7:15752.